TMA-5 synthesis
This is classical synthesis method of TMA-5 was criated with help of experts from BB forum.
A solution of 100 g 1,2,4-trimethoxybenzene in 1 L hexane was cooled to 15 °C and treated with 400 mL of a 15% solution of n-butyllithium in hexane. A white precipitate formed immediately, and stirring was continued for an additional 2 h while the reaction returned to room temperature. There was then added a solution of 40 g freshly distilled propionaldehyde in 100 mL hexane. The reaction was exothermic and, as the stirring was continued, the precipitate gradually dissolved. Stirring was continued overnight at room temperature. There was then added 1 L H2O, and the reaction was acidified with HCl. The hexane phase was separated, and the remaining aqueous phase was extracted with hexane, then with Et2O. The pooled organic extracts were stripped of solvent under vacuum, and the residue distilled to give 60 g ethyl 2,3,6-trimethoxyphenyl carbinol, with an index of refraction nD20 = 1.5192. From the Et2O extracts above, additional carbinol was obtained, containing a small amount of the starting 1,2,4-trimethoxybenzene. The two materials were readily separated by vacuum distillation, providing an additional 21 g of carbinol.
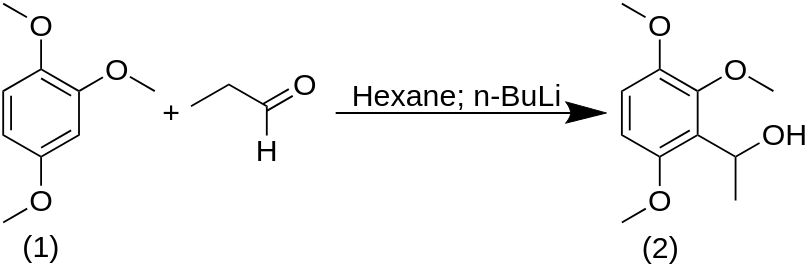
The above alcohol, 60 g of ethyl 2,3,6-trimethoxyphenyl carbinol, was stirred without solvent and cooled to 0 °C with an external ice bath. There was then added 80 g PBr3 at a rate that maintained the temperature below 60 °C. At the end of the addition, there were added quantities of chipped ice, followed by H2O. The reaction mixture was extracted with 3×100 mL Et2O, and removal of the solvent provided 60 g of 1-bromo-1-(2,3,6-trimethoxyphenyl)propane which was used in the following dehydrobromination step without further purification.
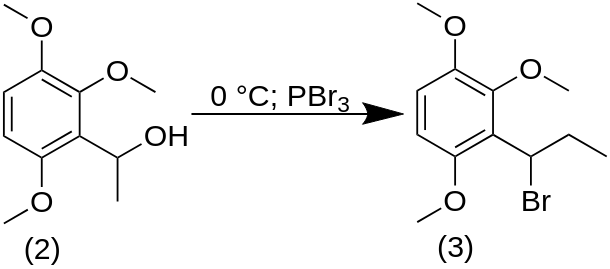
A solution of the above 60 g of 1-bromo-1-(2,3,6-trimethoxyphenyl)propane in an equal weight of EtOH was treated with 120 g of flaked KOH. The exothermic reaction was allowed to run its course with stirring continued overnight. The mixture was then quenched in H2O and extracted with 3×200 mL CH2Cl2. Removal of the solvent from the pooled extracts gave a crude product which contained no starting bromo material, but which was contaminated with an appreciable quantity of the ethoxy analogue, 1-ethoxy-1-(2,3,6-trimethoxyphenyl)propane. This impure product was heated briefly to 80 °C with 50% H2SO4. Cooling, dilution with water, and re-extraction with 3×100 mL CH2Cl2 gave, after removal of the volatiles under vacuum, 1-(2,3,6-trimethoxyphenyl)propene. This was distilled to provide 7.0 g of a clear oil that was a 12:1 ratio of the trans- and cis-isomers.
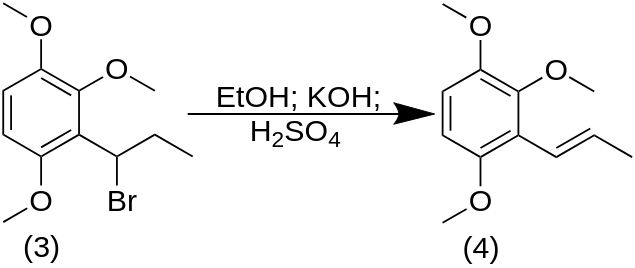
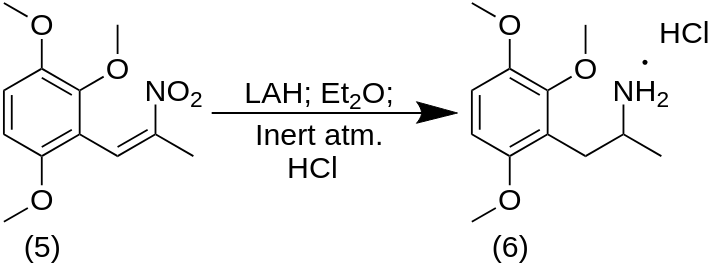
A well-stirred solution of 6.8 g of the mixed isomers of 1-(2,3,6-trimethoxyphenyl)propene in 40 g of dry acetone was treated with 3.2 g pyridine and cooled to 0 °C with an external ice bath. There was then added 6.5 g tetranitromethane over the course of 1 min, the stirring was continued for an additional 2 min, and then the reaction mixture was quenched by the addition of 2.2 g KOH in 40 mL H2O. There was additional H2O added, and the organics were extracted with 3×75 mL CH2Cl2. The solvent from the pooled extracts was removed under vacuum, and the 5.3 g residue distilled at 0.2 mm/Hg. A fraction boiling at 150-170 °C proved to be largely 2,3,6-trimethoxybenzaldehyde. A second fraction (170-200 °C at 0.2 mm/Hg) also spontaneously crystallized to a yellow solid. This was recrystallized from MeOH to provide, after drying to constant weight, 2.8 g of 2-nitro-1-(2,3,6-trimethoxyphenyl)propene with a m.p. of 73-74 °C.
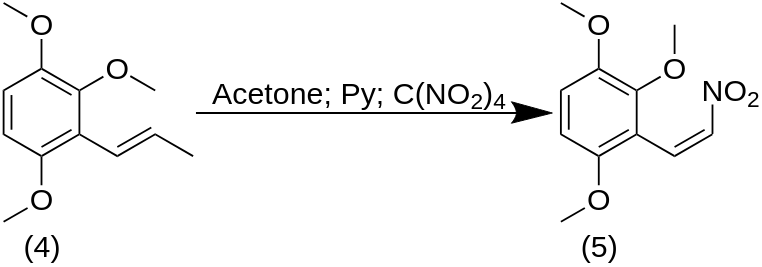
To a refluxing and stirred suspension of 2.4 g LAH in 300 mL anhydrous Et2O and under an inert atmosphere, there was added a solution of 2.4 g 2-nitro-1-(2,3,6-trimethoxyphenyl)propene in 100 mL anhydrous Et2O. The mixture was held at reflux for 4 h, cooled, and then the excess hydride cautiously destroyed by the addition of 1.5 N H2SO4. There was then added 40 g potassium sodium tartrate followed by sufficient aqueous NaOH to raise the pH to >9. The Et2O phase was separated, and the remaining aqueous phase extracted with 3×100 mL CH2Cl2. The organic phase and extracts were combined, and the solvent removed under vacuum yielding 1.8 g of a colorless oil. This was dissolved in 200 mL anhydrous Et2O which was saturated with anhydrous HCl gas. There was generated a thick oil that slowly crystallized. The resulting white crystalline solid was removed by filtration, providing 2.2 g 2,3,6-trimethoxyamphetamine hydrochloride (TMA-5). The m.p. was 124-125 °C.